Prion Molecule -
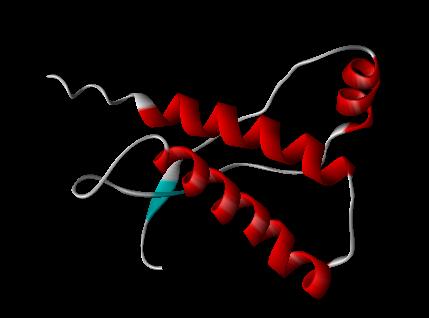
PRION PROTEIN 1AG2 --(showing fragments 121-231)
PDB FILE: 1AG2 PRION PROTEIN, BRAIN, GLYCOPROTEIN, GPI-ANCHOR
To View the Prion 3D using Jsmol
A prion short for proteinaceous infectious particle (-on by analogy to virion) is an infectious agent which according to the "protein only" hypothesis is composed only of protein.[2] Prions cause a number of diseases in a variety of mammals, including bovine spongiform encephalopathy (BSE aka "mad cow disease") in cattle and Creutzfeldt-Jakob disease (CJD) in humans. All known prion diseases affect the structure of the brain or other neural tissue, and all are currently untreatable and fatal.[3] In general usage, prion can refer to both the theoretical unit of infection or the specific protein (e.g. PrP) that is thought to be the infective agent, whether or not it is in an infective state.
Prions are believed to infect and propagate by refolding abnormally into a structure which is able to convert normal molecules of the protein into the abnormally structured form. All known prions induce the formation of an amyloid fold, in which the protein polymerises into an aggregate consisting of tightly packed beta sheets. This altered structure is extremely stable and accumulates in infected tissue, causing cell death and tissue damage.[4]This stability means that prions are resistant to denaturation by chemical and physical agents, making disposal and containment of these particles difficult.
Proteins showing prion behaviour are also found in some fungi and have been very important in helping to understand mammalian prions. However, fungal prions do not appear to cause disease in their hosts and may even confer an evolutionary advantage through a form of protein-based inheritance.[5]
Discovery
Radiation biologist Tikvah Alper and mathematician John Stanley Griffith developed the theory in the 1960s that some transmissible spongiform encephalopathies are caused by an infectious agent made solely of protein.[6][7]This theory was developed to explain the discovery that the mysterious infectious agent causing the diseases scrapie and Creutzfeldt-Jakob Disease resisted ultraviolet radiation (which breaks down nucleic acids - present in viruses and all living things) yet responded to agents that disrupt proteins.
Francis Crick recognized the potential importance of the Griffith protein-only hypothesis for scrapie propagation in the second edition of his famous "Central dogma of molecular biology".[8] While asserting that the flow of sequence information from protein to protein, or from protein to RNA and DNA was "precluded" by the dogma, he noted that Griffith's hypothesis was a potential difficulty (although it was not so promoted by Griffith). As the revised "dogma" was formulated, in part, to accommodate the then recent discovery of reverse transcription by Howard Temin and David_Baltimore (Nobel Prize, 1975), proof of the protein-only hypothesis might have been seen as a sure bet for a future Prize.
Stanley B. Prusiner of the University of California, San Francisco announced in 1982 that his team had purified infectious material and that the infectious agent consisted mainly of a specific protein, although they had not managed to satisfactorily isolate the protein until two years after making his announcement.[9] Prusiner coined the word "prion" as a name for the infectious agent, by combining the first two syllables of the words proteinaceousand infectious.[10] While the infectious agent was named a prion, the specific protein that the prion was made of was named PrP, an abbreviation for "protease-resistant protein". Prusiner was awarded the Nobel Prize in Physiology or Medicine in 1997 for his research into prions.[11]
Structure
Isoforms
The protein that prions are made of is found throughout the body, even in healthy people and animals. However, the prion protein found in infectious material has a different structure and is resistant to proteases, the enzymes in the body that can normally break down proteins. The normal form of the protein is called PrPC, while the infectious form is called PrPSc ” the C refers to 'cellular' or 'common' PrP, while the Sc refers to 'scrapie', a prion disease occurring in sheep.[12] While PrPC is structurally well-defined, PrPSc is certainly polydisperse and defined at a relatively poor level. PrP can be induced to fold into other more-or-less well-defined isoforms in vitro, and their relationship to the form(s) that are pathogenic in vivo is not yet clear.
PrPC
PrPC is a normal protein found on the membranes of cells. It has 209 amino acids (in humans), one disulphide bond, a molecular weight of 35-36kDa and a mainly alpha-helical structure. Several topological forms exist; one cell surface form anchored via glycolipid and two transmembrane forms.[13] Its function has not been fully resolved. PrPC binds copper (II) ions with high affinity..[14] The significance of this is not clear, but it presumably relates to PrP structure or function. PrPC is readily digested by proteinase K and can be liberated from the cell surface in vitro by the enzyme phosphoinositide phospholipase C (PI-PLC), which cleaves the glycophosphatidylinositol (GPI) glycolipid anchor.[15]
PrPSc
The infectious isoform of PrPC, known as PrPSc, is able to convert normal PrPC proteins into the infectious isoform by changing their conformation. Although the exact 3D structure of PrPSc is not known, there is increased β-sheet content in the diseased form of the molecule, replacing normal areas of α-helix.[16] Aggregations of these abnormal isoforms may form highly structured amyloid fibers. The end of a fiber acts as a template for the free protein molecules, causing the fiber to grow. Small differences in the amino acid sequence of prion-forming regions lead to distinct structural features on the surface of prion fibers. As a result, only free protein molecules that are identical in amino acid sequence to the prion protein can be recruited into the growing fiber.
Function
PrP and long-term memory
There is evidence that PrP may have a normal function in maintenance of long term memory.[17] Maglio and colleagues have shown that mice without the genes for normal cellular PrP protein have altered hippocampal LTP.[18]
Prion disease
Prions cause neurodegenerative disease by aggregating extracellularly within the central nervous system to form plaques known as amyloids, which disrupt the normal tissue structure. This disruption is characterized by "holes" in the tissue with resultant spongy architecture due to the vacuole formation in the neurons.[19] Other histological changes include astrogliosis and the absence of an inflammatory reaction.[20] While the incubation period for prion diseases is generally quite long, once symptoms appear the disease progresses rapidly, leading to brain damage and death.[21] Neurodegenerative symptoms can include convulsions, dementia, ataxia (balance and coordination dysfunction), and behavioural or personality changes.
All known prion diseases, collectively called transmissible spongiform encephalopathies (TSEs), are untreatable and fatal.[22] However, a vaccine has been developed in mice that may provide insight into providing a vaccine in humans to resist prion infections.[23] Additionally, in 2006 scientists announced that they had genetically engineered cattle lacking a necessary gene for prion production - thus theoretically making them immune to BSE,[24] building on research indicating that mice lacking normally-occurring prion protein are resistant to infection by scrapie prion protein.[25]
Many different mammalian species can be affected by prion diseases, as the prion protein (PrP) is very similar in all mammals.[26] Due to small differences in PrP between different species, it is unusual for a prion disease to be transmitted from one species to another. However, the human prion disease variant Creutzfeldt-Jakob disease is believed to be caused by a prion which typically infects cattle and is transmitted through infected meat.[27]
Some researchers have suggested that metal ion interactions with prion proteins might be relevant to the progression of prion-mediated disease, based on epidemiological studies of clusters of prion disease in locales with low soil concentrations of copper.[28]
In humans:
- Creutzfeldt-Jakob disease (CJD)[29] and its varieties: iatrogenic Creutzfeldt-Jakob disease (iCJD), variant Creutzfeldt-Jakob disease (vCJD), familial Creutzfeldt-Jakob disease (fCJD), and sporadic Creutzfeldt-Jakob disease (sCJD)
- Gerstmann-Sträussler-Scheinker syndrome (GSS)[29]
- Fatal familial insomnia (fFI)[29]
- Sporadic fatal insomnia (sFI)[31]
- Kuru[29]
Although the identity and general properties of prions are now well understood, the mechanism of prion infection and propagation remains mysterious. It is often assumed that the diseased form directly interacts with the normal form to make it rearrange its structure. One idea, the "Protein X" hypothesis, is that an as-yet unidentified cellular protein (Protein X) enables the conversion of PrPC to PrPSc by bringing a molecule of each of the two together into a complex.[32]
Current research suggests that the primary method of infection in animals is through ingestion. It is thought that prions may be deposited in the environment through the remains of dead animals and via urine, saliva, and other body fluids. They may then linger in the soil by binding to clay and other minerals.[33]
Sterilization
Infectious particles possessing nucleic acid are dependent upon it to direct their continued replication. Prions however, are infectious by their effect on normal versions of the protein. Therefore, sterilizing prions involves the denaturation of the protein to a state where the molecule is no longer able to induce the abnormal folding of normal proteins. However, prions are generally quite resistant to denaturation by proteases, heat, radiation, and formalin treatments,[34] although their infectivity can be reduced by such treatments.
Prions can be denatured by subjecting them to a temperatures of 134 degrees Celsius (274 degrees Fahrenheit) for 18 minutes in a pressurised steam autoclave.[35] Ozone sterilization is currently being studied as a potential method for prion deactivation.[36] Renaturation of a completely denatured prion to infectious status has not yet been achieved, however partially denatured prions can be renatured to an infective status under certain artificial conditions.[37]
The World Health Organisation recommends the following procedure for the sterilisation of heat resistant surgical instruments which are potentially contaminated with prions:
(1) Immerse in a pan containing 1N NaOH and heat in a gravity-displacement autoclave at 121°C for 30 min; clean; rinse in water; and subject to routine sterilization
(2) Immerse in 1N NaOH or sodium hypochlorite (20,000 parts per million available chlorine) for 1 h; transfer instruments to water; heat in a gravity-displacement autoclave at 121°C for 1 h; clean; and subject to routine sterilization
(3) Immerse in 1N NaOH or sodium hypochlorite (20,000 parts per million available chlorine) for 1 h; remove and rinse in water, then transfer to an open pan and heat in a gravity-displacement (121°C) or in a porous-load (134°C) autoclave for 1 h; clean; and subject to routine sterilization [38]
Protein-only hypothesis
Prior to the discovery of prions, it was thought that all pathogens used nucleic acids to direct their replication. The "protein-only hypothesis" states that a protein structure can replicate without the use of nucleic acid. This was initially controversial as it contradicts the so-called "central dogma of molecular biology," which describes nucleic acid as the central form of replicative information.
Evidence in favor of a protein-only hypothesis include:[39]
- No virus particles have been conclusively associated with prion diseases
- No nucleic acid has been conclusively associated with infectivity; agent is resistant to degradation by nucleases
- No immune response to infection
- PrPSc experimentally transmitted between one species and another results in PrPSc with the amino-acid sequence of the recipient species, suggesting that replication of the donor agent does not occur
- Level of infectivity is associated with levels of PrPSc
- PrPSc and PrPC do not differ in amino-acid sequence, therefore a PrPSc-specific nucleic acid is a redundant concept
- Familial prion disease occurs in families with a mutation in the PrP gene, and mice with PrP mutations develop prion disease despite controlled conditions where transmission is prevented
Viral hypothesis
The protein-only hypothesis was initially met with skepticism and still has critics. For more than a decade, Yale University neuropathologist Laura Manuelidis has been proposing that prion diseases are caused instead by an unidentifiable "slow virus". In January 2007 she and her colleagues published an article in the Proceedings of the National Academy of Science reporting to have found the virus in 10%, or less, of their scrapie-infected cells in culture.[40][41]
The Virion Hypothesis states that TSEs are caused by a replicable informational molecule (which is likely to be a nucleic acid) bound to PrP. Many TSEs, including Scrapie and BSE, show strains with specific and distinct biological properties, a feature which supporters of the Virion hypothesis feel is not explained by prions. The presence of a nucleic acid bound to the protein would explain the strains observed. It has also been shown that TSEs including BSE retain their host-specific properties after passage through many different species. [42]
Evidence in favor of a viral hypothesis include:[39]
- No bacteria or other living organisms have been found in prion-affected organisms, defaulting to the idea that a virus must be involved
- The long incubation and rapid onset of symptoms resembles some viral infections, such as HIV-induced AIDS
- Differences in prion infectivity, incubation, symptomology and progression among species resembles the "strain variation" seen between viruses, especially RNA viruses
What Causes Mad Cows Disease?
Bovine spongiform encephalopathy (BSE), commonly known as mad cow disease, is a transmissible spongiform encephalopathy and fatal neurodegenerative disease in cattle that may be passed to humans who have eaten infected flesh. When it has been transmitted to humans, it is known as new Variant Creutzfeldt–Jakob disease (vCJD or nvCJD), and is now believed to be caused by the prion molecule. The molecular basis of prion replication is a change in conformation of the normal cellular prion protein (PrPC) into the abnormal and misfolded conformer (PrPSc) that is partially protease-resistant (PrPres).
..."PrPc is a normal cell-surface glycoprotein that is conformationally characterized by two alpha helices and two complex-type N-linked oligosaccharide chains. This protein is unique in its propensity to misfold into a neurodegenerative disease-causing proteinaceous infectious particle, known as a prion. PrPc can undergo conversion into PrPSc through spontaneous misfolding, a genetic mutation of the human PRNP gene, or exposure to a prion from an external source. When this happens, its composition shifts from an alpha-helical, soluble protein low in beta pleated sheets to an insoluble, protease-resistant particle with a high percentage of beta sheets (see The shape of infectious prions). This resulting misfolded form, known formally as PrPSc, is pathogenic and aggregates in neural tissue by recruiting and converting more PrPc to PrPSc, ultimately causing neuronal cell dysfunction followed by cell death. .." See Reference 44.
Prion infections are extremely rare in humans, in whom approximately 99% of all cases occur sporadically or are inherited because of mutations in the gene encoding PrPC. In these ill- nesses, PrPC misfolds spontaneously without the need for contact with exogenous PrPSc. When expressed in transgenic mice, PrPC molecules with certain genetic prion disease-associated mutations cause neurological syndromes that recapitulate key features of the corresponding human disorders (45).
Alpha to Beta Sheet Conversion
REFERENCES
- "Prion". Oxford English Dictionary. Oxford University Press. 2nd ed. 1989.
- http://www.pnas.org/cgi/content/extract/105/1/11?etoc
- Prusiner SB (1998). "Prions". Proc. Natl. Acad. Sci. USA 95 (23): 13363-83. doi:10.1073/pnas.95.23.13363. PMID 9811807.
- Dobson CM (2001). "The structural basis of protein folding and its links with human disease". Philos Trans R Soc Lond B Biol Sci 356 (1406): 133-145. doi:10.1098/rstb.2000.0758. PMID 11260793.
- Lindquist S, Krobitsch S, Li L and Sondheimer N (2001). "Investigating protein conformation-based inheritance and disease in yeast". Philos Trans R Soc Lond B Biol Sci 356 (1406): 169-176. doi:10.1098/rstb.2000.0762. PMID 11260797.
- Alper T, Cramp W, Haig D, Clarke M (1967). "Does the agent of scrapie replicate without nucleic acid?". Nature 214 (5090): 764-6. doi:10.1038/214764a0. PMID 4963878.
- Griffith J (1967). "Self-replication and scrapie". Nature 215 (5105): 1043-4. doi:10.1038/2151043a0.
- Crick F (1970). "Central dogma of molecular biology". Nature 227 (5258): 561-“3. PMID 43914.
- Taubes, Gary (December 1986), "The game of name is fame. But is it science?", Discover 7 (12): 28-41
- Prusiner, SB (1982). "Novel proteinaceous infectious particles cause scrapie". Science 216 (4542): 136-144. doi:10.1126/science.278.5336.245.
- The Nobel Prize in Physiology or Medicine 1997. NobelPrize.org.
- Priola, Suzette A.; Chesebro, Bruce; Caughey, Byron (2003). "A View from the Top--Prion Diseases from 10,000 Feet". Science 300 (5621): 917-919.
- Hegde RS, Mastrianni JA, Scott MR, Defea KA, Tremblay P, Torchia M, DeArmond SJ, Prusiner SB, Lingappa VR (1998). "A transmembrane form of the prion protein in neurodegenerative disease". Science 276: 827-834. doi:10.1126/science.279.5352.827.
- Hornshaw MP, McDermott JR, Candy JM (1995). "Copper binding to the N-terminal tandem repeat regions of mammalian and avian prion protein". Biochem Biophys Res Commun 207: 621-629. doi:10.1006/bbrc.1995.1233.
- Weissmann, C (2004). "The State of the Prion". Nature Reviews Microbiology 2: 861-871. doi:10.1038/nrmicro1025.
- (1993 December 1) "Conversion of alpha-helices into beta-sheets features in the formation of scrapie prion protein". PNAS USA 90 (23): 10962-6.
- Shorter J, Lindquist S (2005). "Prions as adaptive conduits of memory and inheritance". Nat Rev Genet 6 (6): 435-50. doi:10.1038/nrg1616.
- Maglio L, Perez M, Martins V, Brentani R, Ramirez O (2004). "Hippocampal synaptic plasticity in mice devoid of cellular prion protein". Brain Res Mol Brain Res 131 (1-2): 58-64. doi:10.1016/j.molbrainres.2004.08.004.
- Cotran; Kumar, Collins. Robbins Pathologic Basis of Disease. Philadelphia: W.B Saunders Company. 0-7216-7335-X.
- Belay E. (1999). "Transmissible Spongiform Encephalopathies in Humans". Annu. Rev. Microbiol. 53: 283-314. doi:10.1146/annurev.micro.53.1.283.
- Prion Diseases. US Centers for Disease Control.
- Gilch, Sabine, et al. (2001). "Intracellular re-routing of prion protein prevents propagation of PrPSc and delays onset of prion disease". The EMBO Journal 20 (15): 3957-3966. doi:10.1093/emboj/20.15.3957.
- New York University Medical Center and School of Medicine (2005-05-14). Active Vaccine Prevents Mice From Developing Prion Disease. Science Daily
- Weiss, Rick. "Scientists Announce Mad Cow Breakthrough", The Washington Post, 2007-01-01.Beler H, Aguzzi A, Sailer A, Greiner R, Autenried P, Aguet M, Weissmann C (1993). "Mice devoid of PrP are resistant to scrapie". Cell 73 (7): 1339-47. doi:10.1016/0092-8674(93)90360-3. PMID 8100741.
- Collinge J (2001). "Prion diseases of humans and animals: their causes and molecular basis". Annu Rev Neurosci 24: 519-50. doi:10.1146/annurev.neuro.24.1.519. PMID 11283320.
- Ironside, JW (2006). "Variant Creutzfeldt-Jakob disease: risk of transmission by blood transfusion and blood therapies". Haemophilia 12 (s1): 8-15. doi:10.1111/j.1365-2516.2006.01195.x. PMID 16445812.
- 2000-09-22, Normal Function of Prions, Statement to the BSE Inquiry (PDF).
- "90. Prions - ICTVdB Index of Viruses." (Website.) U.S. National Institutes of Health website.
- Hussein, Mansour F. and Saud I. Al-Mufarrej. (2004.) "Prion Diseases: A Review; II. Prion Diseases in Man and Animals." Scientific Journal of King Faisal University (Basic and Applied Sciences), vol. 5, no. 2 1425, p. 139.
- (1999-.) "BSE proteins may cause fatal insomnia." (News website.) BBC News.
- Telling G, Scott M, Mastrianni J, Gabizon R, Torchia M, Cohen F, DeArmond S, Prusiner S (1995). "Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein". Cell 83 (1): 79-90.
- Johnson C, Pedersen J, Chappell R, McKenzie D, Aiken J (2007). "Oral Transmissibility of Prion Disease is Enhanced by Binding to Soil Particles". PLoS Pathogens 3 (7). doi:10.1371/journal.ppat.0030093.
- Qin K, O'Donnell M, Zhao R (2006). "Doppel: more rival than double to prion". Neuroscience 141 (1): 1-8. doi:10.1016/j.neuroscience.2006.04.057.
- Collins SJ, Lawson VA, Masters CL (2004). "Transmissible spongiform encephalopathies". Lancet 363 (9402): 51-61. doi:10.1016/S0140-6736(03)15171-9.
- Ozone Sterilization - UK Health Protection Agency
- Weissmann C, Enari M, Klöhn PC, Rossi D, Flechsig E (2002). "Transmission of prions". Proc. Natl. Acad. Sci. U.S.A. 99 Suppl 4: 16378-83. :10.1073/pnas.172403799.
- Sutton JM, Dickinson J, Walker JT and Raven NDH (2006). "Methods to Minimize the Risks of Creutzfeldt-Jakob Disease by Surgical Proc.:10.1086/507030.
- Baker & Ridley (1996). Prion Disease. New Jersey: Humana Press. 0-89603-342-2.
- "Pathogenic Virus Found in Mad Cow Cells", Yale, February 2.
- Laura Manuelidis, Zhoa-Xue Yu, Nuria Barquero, and Brian Mullins (February 6, 2007). "Cells infected with scrapie and Creutzfeldt–Jakob disease agents produce intracellular 25-nm virus-like particles". Proceedings of the National Academy of Sciences of the United States of America 104 (6): 1965-1970. doi:10.1073/pnas.0610999104.
- Farquhar C, Somerville R and Bruce M (1998). "Straining the prion hypothesis". Nature 391: 345-346. doi:10.1038/34818.
- 2000-09-22, Normal Function of Prions, Statement to the BSE Inquiry
- Oesch B, Westaway D, Wälchli M, McKinley M, Kent S, Aebersold R, Barry R, Tempst P, Teplow D, Hood L (1985). "A cellular gene encodes scrapie PrP 27-30 protein". Cell 40 (4): 735-46.
- Ashley Howard, Shulammite Lim. Priya Lakshman, Priya Lakshmanalse and Cheyenne Loo (2018) PrPc The Normal Prion. FASEB Journal 20 April 2018.
- Roberto Chiesa (2015) The Elusive Role of the Prion Protein and the Mechanism of Toxicity in Prion Disease. Plos Pathogens
Molecules of Life Resources